
}}
}}
”’Paluratide”’ (development code ”’LUNA18”’) was an investigational [[cyclic peptide]] [[KRAS|KRAS inhibitor]] developed by [[Chugai Pharmaceutical]], a member of the [[Roche]] Group, for the treatment of cancers with [[KRAS]] mutations.<ref name=”Tanada2023“>{{cite journal |last1=Tanada |first1=Mikimasa |last2=Tamiya |first2=Minoru |last3=Matsuo |first3=Atsushi |display-authors=etal |title=Development of Orally Bioavailable Peptides Targeting an Intracellular Protein: From a Hit to a Clinical KRAS Inhibitor |journal=Journal of the American Chemical Society |date=2023 |volume=145 |issue=30 |pages=16610–16620 |doi=10.1021/jacs.3c03886 |pmid=37463267 |bibcode=2023JAChS.14516610T |url=https://pubs.acs.org/doi/10.1021/jacs.3c03886|url-access=subscription }}</ref> The compound was notable as an orally bioavailable macrocyclic peptide that could target intracellular [[protein-protein interaction]]s, a class of targets traditionally considered “undruggable.”<ref name=”Ohta2023“>{{cite journal |last1=Ohta |first1=Atsushi |last2=Tanada |first2=Mikimasa |last3=Shinohara |first3=Shojiro |display-authors=etal |title=Validation of a New Methodology to Create Oral Drugs beyond the Rule of 5 for Intracellular Tough Targets |journal=Journal of the American Chemical Society |date=2023 |volume=145 |issue=44 |pages=24035–24051 |doi=10.1021/jacs.3c07145 |pmid=37874670 |bibcode=2023JAChS.14524035O |url=https://pubs.acs.org/doi/10.1021/jacs.3c07145|url-access=subscription }}</ref>
”’Paluratide”’ (development code ”’LUNA18”’) was an investigational [[cyclic peptide]] [[KRAS|KRAS inhibitor]] developed by [[Chugai Pharmaceutical]], a member of the [[Roche]] Group, for the treatment of cancers with [[KRAS]] mutations.<ref name=””>{{cite journal |=Tanada Tamiya Matsuo |title=Development of Orally Bioavailable Peptides Targeting an Intracellular Protein: From a Hit to a Clinical KRAS Inhibitor |journal=Journal of the American Chemical Society | volume=145 |issue=30 |pages=16610–16620 |= |pmid=37463267 |= 10.1021/jacs.3c03886|= }}</ref> The compound was notable as an orally bioavailable macrocyclic peptide that could target intracellular [[protein-protein interaction]]s, a class of targets traditionally considered “undruggable.”<ref name=””>{{cite journal |=Ohta Tanada Shinohara |title=Validation of a New Methodology to Create Oral Drugs beyond the Rule of 5 for Intracellular Tough Targets |journal=Journal of the American Chemical Society | volume=145 |issue=44 |pages=24035–24051 |= |pmid=37874670 |= 10.1021/jacs.3c07145|= }}</ref>
Development was discontinued in July 2025 due to a narrow therapeutic window compared to competing KRAS inhibitors.<ref name=”Fierce2025″>{{cite web |title=Roche axes 4 Chugai solid tumor assets in early-phase clear-out |url=https://www.fiercebiotech.com/biotech/roche-axes-4-chugai-solid-tumor-assets-early-phase-clearout |website=Fierce Biotech |date=October 24, 2025}}</ref>
Development was discontinued in July 2025 due to a narrow therapeutic window compared to competing KRAS inhibitors.<ref name=”Fierce2025″>{{cite web |title=Roche axes 4 Chugai solid tumor assets in early-phase clear-out |url=https://www.fiercebiotech.com/biotech/roche-axes-4-chugai-solid-tumor-assets-early-phase-clearout | = }}</ref>
== Mechanism of action ==
== Mechanism of action ==
Paluratide functions as a pan-RAS inhibitor, targeting multiple RAS isoforms including [[KRAS]], [[Neuroblastoma RAS viral oncogene homolog|NRAS]], and [[HRAS]].<ref name=”Tanada2023” /> The compound binds with high affinity to KRAS<sup>G12D</sup>, with a dissociation constant (K<sub>d</sub>) of 0.043 nM, and blocks the interaction between KRAS<sup>G12D</sup> and the [[Son of sevenless|guanine nucleotide exchange factor]] [[SOS1]] with an [[IC50|IC<sub>50</sub>]] of less than 2.2 nM.<ref name=”MedChem”>{{cite web |title=LUNA18 (Paluratide) – KRAS Inhibitor, ERK Inhibitor, RAS Inhibitor |url=https://www.medchemexpress.com/luna18.html |website=MedChemExpress}}</ref>
Paluratide functions as a pan-RAS inhibitor, targeting multiple RAS isoforms including [[KRAS]], [[Neuroblastoma RAS viral oncogene homolog|NRAS]], and [[HRAS]].<ref name=”” /> The compound binds with high affinity to KRAS<sup>G12D</sup>, with a dissociation constant (K<sub>d</sub>) of 0.043 nM, and blocks the interaction between KRAS<sup>G12D</sup> and the [[Son of sevenless|guanine nucleotide exchange factor]] [[SOS1]] with an [[IC50|IC<sub>50</sub>]] of less than 2.2 nM.<ref name=”MedChem”>{{cite web |title=LUNA18 (Paluratide) – KRAS Inhibitor, ERK Inhibitor, RAS Inhibitor |url=https://www.medchemexpress.com/luna18.html |website=MedChemExpress}}</ref>
Unlike covalent KRAS inhibitors that target specific mutations (such as [[sotorasib]] for KRAS<sup>G12C</sup>), paluratide was designed to inhibit RAS proteins through disruption of protein-protein interactions with [[guanine nucleotide exchange factors]] (GEFs).<ref name=”Tanada2023” /> This mechanism allows the drug to affect RAS signalling regardless of the specific mutation, theoretically providing broader applicability across different KRAS-mutant cancers. The compound also demonstrates activity against downstream signalling pathways, affecting [[Extracellular signal-regulated kinases|ERK]] and [[Protein kinase B|AKT]] phosphorylation.<ref name=”MedChem” />
Unlike covalent KRAS inhibitors that target specific mutations (such as [[sotorasib]] for KRAS<sup>G12C</sup>), paluratide was designed to inhibit RAS proteins through disruption of protein-protein interactions with [[guanine nucleotide exchange factors]] (GEFs).<ref name=”” /> This mechanism allows the drug to affect RAS signalling regardless of the specific mutation, theoretically providing broader applicability across different KRAS-mutant cancers. The compound also demonstrates activity against downstream signalling pathways, affecting [[Extracellular signal-regulated kinases|ERK]] and [[Protein kinase B|AKT]] phosphorylation.<ref name=”MedChem” />
== Medical uses ==
== Medical uses ==
Paluratide was being developed for the treatment of locally advanced or metastatic [[solid tumor]]s harbouring [[Ras GTPase|RAS]] gene alterations.<ref name=”NCT05012618″>{{cite journal |title=A Dose-escalation Study of LUNA18 in Patients With Locally Advanced or Metastatic Solid Tumors (With Expansion). |url=https://clinicaltrials.gov/study/NCT05012618 |website=ClinicalTrials.gov |date=29 July 2025 |id=NCT05012618}}</ref> The drug demonstrated significant cellular activity against multiple cancer types with KRAS mutations in preclinical studies, including [[colorectal cancer]], [[gastric cancer]], [[non-small cell lung cancer]], and [[pancreatic cancer]].<ref name=”Tanada2023” />
Paluratide was being developed for the treatment of locally advanced or metastatic [[solid tumor]]s harbouring [[Ras GTPase|RAS]] gene alterations.<ref name=”NCT05012618″>{{cite journal |title=A Dose-escalation Study of LUNA18 in Patients With Locally Advanced or Metastatic Solid Tumors (With Expansion). |url=https://clinicaltrials.gov/study/NCT05012618 |website=ClinicalTrials.gov | NCT05012618}}</ref> The drug demonstrated significant cellular activity against multiple cancer types with KRAS mutations in preclinical studies, including [[colorectal cancer]], [[gastric cancer]], [[non-small cell lung cancer]], and [[pancreatic cancer]].<ref name=”” />
== Chemistry ==
== Chemistry ==
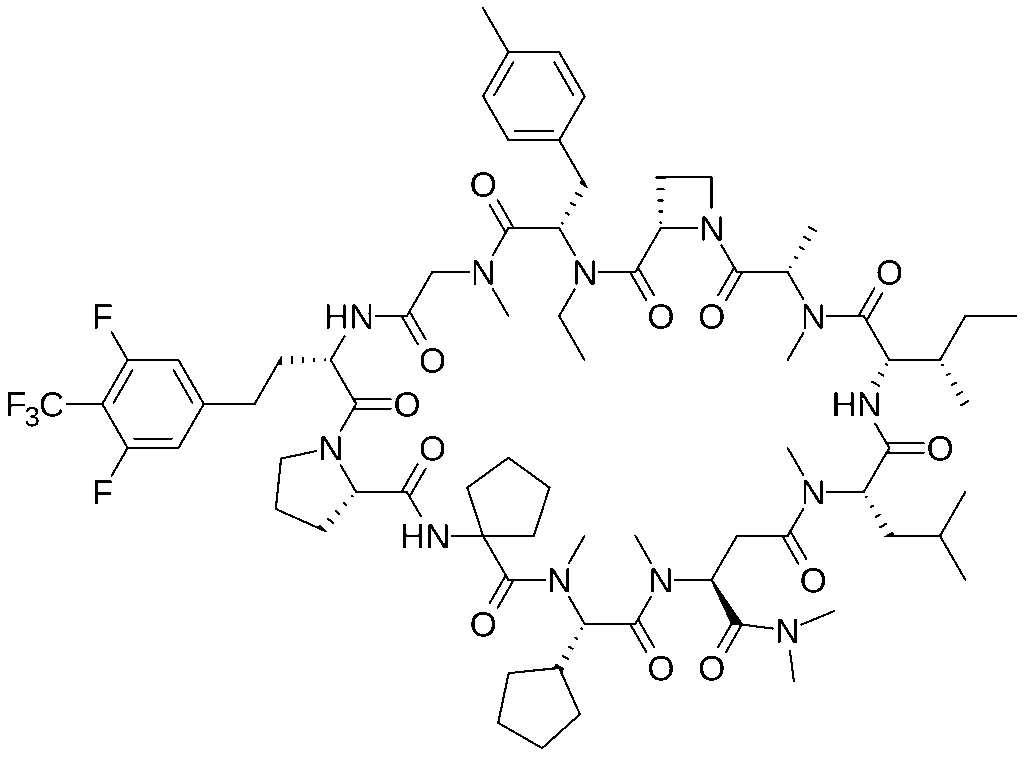
Paluratide is an 11-member (11-mer) [[cyclic peptide]] with a molecular weight in the range of 1000–2000 g/mol, classified as a “middle-size” cyclic peptide.<ref name=”Tanada2023” /> The compound features extensive N-alkylation, a modification that reduces hydrogen bond donors and improves oral absorption while maintaining cellular permeability.<ref name=”Ohta2023” /> Its structure allows it to navigate the challenging boundary between small molecules and biologics, achieving properties of both classes. The compound demonstrated oral bioavailability ranging from 21% to 47% in preclinical animal studies without requiring special formulations.<ref name=”Tanada2023” />
Paluratide is an 11-member (11-mer) [[cyclic peptide]] with a molecular weight in the range of 1000–2000 g/mol, classified as a “middle-size” cyclic peptide.<ref name=”” /> The compound features extensive N-alkylation, a modification that reduces hydrogen bond donors and improves oral absorption while maintaining cellular permeability.<ref name=”” /> Its structure allows it to navigate the challenging boundary between small molecules and biologics, achieving properties of both classes. The compound demonstrated oral bioavailability ranging from 21% to 47% in preclinical animal studies without requiring special formulations.<ref name=”” />
== Discovery ==
== Discovery ==
Paluratide was discovered through Chugai Pharmaceutical’s cyclic peptide platform using an [[mRNA display]] library screening approach.<ref name=”Tanada2023” /> The initial hit compound, designated AP8747, was identified from the mRNA display library and subsequently underwent extensive chemical optimization without scaffold hopping (maintaining the basic cyclic peptide structure).<ref name=”Tanada2023” /> The optimization focused on increasing plasma stability, improving absorption, reducing clearance, and reducing hydrogen bond donors to achieve oral bioavailability.
Paluratide was discovered through Chugai Pharmaceutical’s cyclic peptide platform using an [[mRNA display]] library screening approach.<ref name=”” /> The initial hit compound, designated AP8747, was identified from the mRNA display library and subsequently underwent extensive chemical optimization without scaffold hopping (maintaining the basic cyclic peptide structure).<ref name=”” /> The optimization focused on increasing plasma stability, improving absorption, reducing clearance, and reducing hydrogen bond donors to achieve oral bioavailability.
The final clinical compound, LUNA18, emerged after modifications to four amino acid positions (positions 5, 7, 10, and 11) from an intermediate compound (compound 40). Key structure-activity relationship findings included: the side chain at position 5 preferring aromatic over aliphatic groups; physicochemical properties being adjustable at position 11; and biological activity enhancement through modifications at positions 7 and 10.<ref name=”Tanada2023” />
The final clinical compound, LUNA18, emerged after modifications to four amino acid positions (positions 5, 7, 10, and 11) from an intermediate compound (compound 40). Key structure-activity relationship findings included: the side chain at position 5 preferring aromatic over aliphatic groups; physicochemical properties being adjustable at position 11; and biological activity enhancement through modifications at positions 7 and 10.<ref name=”” />
Chugai also developed a novel synthetic methodology that enabled the broadly applicable synthesis of highly N-alkylated cyclic peptide-like drugs.<ref name=”Nomura2022“>{{cite journal |last1=Nomura |first1=Kenichi |last2=Hashimoto |first2=Satoshi |last3=Takeyama |first3=Ryuuichi |display-authors=etal |date=2022 |title=Broadly Applicable and Comprehensive Synthetic Method for N-Alkyl-Rich Drug-like Cyclic Peptides |url=https://pubs.acs.org/doi/full/10.1021/acs.jmedchem.2c01296 |journal=Journal of Medicinal Chemistry |volume=65 |issue=19 |pages=13401–13412 |doi=10.1021/acs.jmedchem.2c01296 |pmid=36109865|url-access=subscription }}</ref> This method overcame three major technical challenges: formation of [[diketopiperazine]], insufficient reactivity of [[Amide|amidation]] due to steric hindrance, and instability of cyclic peptides under acidic conditions. Using this approach, more than 4,000 cyclic peptides were synthesized with a process yield of 31% and final product purity of 97%.<ref name=”Nomura2022” />
Chugai also developed a novel synthetic methodology that enabled the broadly applicable synthesis of highly N-alkylated cyclic peptide-like drugs.<ref name=””>{{cite journal |=Nomura Hashimoto Takeyama |title=Broadly Applicable and Comprehensive Synthetic Method for N-Alkyl-Rich Drug-like Cyclic Peptides | journal=Journal of Medicinal Chemistry |volume=65 |issue=19 |pages=13401–13412 |doi=10.1021/acs.jmedchem.2c01296 }}</ref> This method overcame three major technical challenges: formation of [[diketopiperazine]], insufficient reactivity of [[Amide|amidation]] due to steric hindrance, and instability of cyclic peptides under acidic conditions. Using this approach, more than 4,000 cyclic peptides were synthesized with a process yield of 31% and final product purity of 97%.<ref name=”” />
== Clinical trials ==
== Clinical trials ==
== Discontinuation ==
== Discontinuation ==
In July 2025, [[Chugai Pharmaceutical Co.|Chugai Pharmaceutical]] announced the discontinuation of paluratide along with four other early-stage clinical development projects (SAIL66, SOF10, STA551, and AMY109).<ref name=”Chugai2025″>{{cite press release |title=Chugai Announces 2025 2nd Quarter Results |publisher=Chugai Pharmaceutical |date=July 24, 2025 |url=https://www.chugai-pharm.co.jp/english/news/detail/20250724160001_1153.html}}</ref> The decision was made to dynamically and strategically allocate resources to priority projects to maximize the success rate of achieving the company’s TOP I 2030 strategic goals.<ref name=”Chugai2025″ />
In July 2025, [[Chugai Pharmaceutical Co.|Chugai Pharmaceutical]] announced the discontinuation of paluratide along with four other early-stage clinical development projects (SAIL66, SOF10, STA551, and AMY109).<ref name=”Chugai2025″>{{cite press release |title=Chugai Announces 2025 2nd Quarter Results | date=July 24, 2025 |url=https://www.chugai-pharm.co.jp/english/news/detail/20250724160001_1153.html}}</ref> The decision was made to dynamically and strategically allocate resources to priority projects to maximize the success rate of achieving the company’s TOP I 2030 strategic goals.<ref name=”Chugai2025″ />
Chugai CEO Osamu Okuda stated on an earnings call in July 2025 that paluratide had a narrower therapeutic window than rival KRAS inhibitor products, leading the company to shift its focus to AUBE00, its second clinical-stage mid-size molecule KRAS inhibitor.<ref name=”Fierce2025″ /> Following Chugai’s announcement, [[Roche]] removed paluratide from its clinical pipeline in October 2025 as part of its third-quarter results update.<ref name=”Fierce2025″ />
Chugai CEO Osamu Okuda stated on an earnings call in July 2025 that paluratide had a narrower therapeutic window than rival KRAS inhibitor products, leading the company to shift its focus to AUBE00, its second clinical-stage mid-size molecule KRAS inhibitor.<ref name=”Fierce2025″ /> Following Chugai’s announcement, [[Roche]] removed paluratide from its clinical pipeline in October 2025 as part of its third-quarter results update.<ref name=”Fierce2025″ />
*[[Protein–protein interaction|Protein-protein interaction]]
*[[Protein–protein interaction|Protein-protein interaction]]
==References==
==References==
{{Reflist}}
{{Reflist}}
Investigational KRAS inhibitor
Pharmaceutical compound
 |
|
| Other names | LUNA18 |
|---|---|
| Routes of administration |
Oral administration |
| Legal status | |
|
|
| CAS Number | |
| PubChem CID | |
| ChemSpider | |
| UNII | |
| Formula | C73H105F5N12O12 |
| Molar mass | 1437.707 g·mol−1 |
| 3D model (JSmol) | |
|
|
|
|
Paluratide (development code LUNA18) was an investigational cyclic peptide KRAS inhibitor developed by Chugai Pharmaceutical, a member of the Roche Group, for the treatment of cancers with KRAS mutations.[1] The compound was notable as an orally bioavailable macrocyclic peptide that could target intracellular protein-protein interactions, a class of targets traditionally considered “undruggable.”[2]
Development was discontinued in July 2025 due to a narrow therapeutic window compared to competing KRAS inhibitors.[3]
Mechanism of action
[edit]
Paluratide functions as a pan-RAS inhibitor, targeting multiple RAS isoforms including KRAS, NRAS, and HRAS.[1] The compound binds with high affinity to KRASG12D, with a dissociation constant (Kd) of 0.043 nM, and blocks the interaction between KRASG12D and the guanine nucleotide exchange factor SOS1 with an IC50 of less than 2.2 nM.[4]
Unlike covalent KRAS inhibitors that target specific mutations (such as sotorasib for KRASG12C), paluratide was designed to inhibit RAS proteins through disruption of protein-protein interactions with guanine nucleotide exchange factors (GEFs).[1] This mechanism allows the drug to affect RAS signalling regardless of the specific mutation, theoretically providing broader applicability across different KRAS-mutant cancers. The compound also demonstrates activity against downstream signalling pathways, affecting ERK and AKT phosphorylation.[4]
Paluratide was being developed for the treatment of locally advanced or metastatic solid tumors harbouring RAS gene alterations.[5] The drug demonstrated significant cellular activity against multiple cancer types with KRAS mutations in preclinical studies, including colorectal cancer, gastric cancer, non-small cell lung cancer, and pancreatic cancer.[1]
Paluratide is an 11-member (11-mer) cyclic peptide with a molecular weight in the range of 1000–2000 g/mol, classified as a “middle-size” cyclic peptide.[1] The compound features extensive N-alkylation, a modification that reduces hydrogen bond donors and improves oral absorption while maintaining cellular permeability.[2] Its structure allows it to navigate the challenging boundary between small molecules and biologics, achieving properties of both classes. The compound demonstrated oral bioavailability ranging from 21% to 47% in preclinical animal studies without requiring special formulations.[1]
Paluratide was discovered through Chugai Pharmaceutical’s cyclic peptide platform using an mRNA display library screening approach.[1] The initial hit compound, designated AP8747, was identified from the mRNA display library and subsequently underwent extensive chemical optimization without scaffold hopping (maintaining the basic cyclic peptide structure).[1] The optimization focused on increasing plasma stability, improving absorption, reducing clearance, and reducing hydrogen bond donors to achieve oral bioavailability.
The final clinical compound, LUNA18, emerged after modifications to four amino acid positions (positions 5, 7, 10, and 11) from an intermediate compound (compound 40). Key structure-activity relationship findings included: the side chain at position 5 preferring aromatic over aliphatic groups; physicochemical properties being adjustable at position 11; and biological activity enhancement through modifications at positions 7 and 10.[1]
Chugai also developed a novel synthetic methodology that enabled the broadly applicable synthesis of highly N-alkylated cyclic peptide-like drugs.[6] This method overcame three major technical challenges: formation of diketopiperazine, insufficient reactivity of amidation due to steric hindrance, and instability of cyclic peptides under acidic conditions. Using this approach, more than 4,000 cyclic peptides were synthesized with a process yield of 31% and final product purity of 97%.[6]
A Phase 1 dose-escalation and cohort expansion study (NCT05012618) was initiated in August 2021 to evaluate the safety, pharmacokinetics, pharmacodynamics, and preliminary activity of paluratide administered as a single agent or in combination with other anti-cancer drugs.[5] The study, in the United States and Japan, was designed to enrol approximately 195 patients with locally advanced or metastatic solid tumors positive for documented RAS alterations.[5]
Paluratide was administered orally as capsules.[5] The study also evaluated combination therapy with cetuximab, an EGFR inhibitor.[5]
In July 2025, Chugai Pharmaceutical announced the discontinuation of paluratide along with four other early-stage clinical development projects (SAIL66, SOF10, STA551, and AMY109).[7] The decision was made to dynamically and strategically allocate resources to priority projects to maximize the success rate of achieving the company’s TOP I 2030 strategic goals.[7]
Chugai CEO Osamu Okuda stated on an earnings call in July 2025 that paluratide had a narrower therapeutic window than rival KRAS inhibitor products, leading the company to shift its focus to AUBE00, its second clinical-stage mid-size molecule KRAS inhibitor.[3] Following Chugai’s announcement, Roche removed paluratide from its clinical pipeline in October 2025 as part of its third-quarter results update.[3]
- ^ a b c d e f g h i Tanada M, Tamiya M, Matsuo A, Chiyoda A, Takano K, Ito T, et al. (August 2023). “Development of Orally Bioavailable Peptides Targeting an Intracellular Protein: From a Hit to a Clinical KRAS Inhibitor”. Journal of the American Chemical Society. 145 (30): 16610–16620. Bibcode:2023JAChS.14516610T. doi:10.1021/jacs.3c03886. PMID 37463267.
- ^ a b Ohta A, Tanada M, Shinohara S, Morita Y, Nakano K, Yamagishi Y, et al. (November 2023). “Validation of a New Methodology to Create Oral Drugs beyond the Rule of 5 for Intracellular Tough Targets”. Journal of the American Chemical Society. 145 (44): 24035–24051. Bibcode:2023JAChS.14524035O. doi:10.1021/jacs.3c07145. PMID 37874670.
- ^ a b c Taylor NP (24 October 2025). “Roche axes 4 Chugai solid tumor assets in early-phase clear-out”. Fierce Biotech.
- ^ a b “LUNA18 (Paluratide) – KRAS Inhibitor, ERK Inhibitor, RAS Inhibitor”. MedChemExpress.
- ^ a b c d e “A Dose-escalation Study of LUNA18 in Patients With Locally Advanced or Metastatic Solid Tumors (With Expansion)”. ClinicalTrials.gov. 29 July 2025. NCT05012618.
- ^ a b Nomura K, Hashimoto S, Takeyama R, Tamiya M, Kato T, Muraoka T, et al. (October 2022). “Broadly Applicable and Comprehensive Synthetic Method for N-Alkyl-Rich Drug-like Cyclic Peptides”. Journal of Medicinal Chemistry. 65 (19): 13401–13412. doi:10.1021/acs.jmedchem.2c01296. PMID 36109865.
- ^ a b “Chugai Announces 2025 2nd Quarter Results” (Press release). Chugai Pharmaceutical. 24 July 2025.

