


Obesity is a multifactorial disease associated with disturbances in energy regulation pathways, feelings of hunger, and food intake that can influence many health consequences.[1] There are three forms of obesity: the most common, polygenic obesity, entails the interaction of many genes and environmental factors. Syndromic and monogenic rare forms of obesity can be differentiated based on the presence or absence of associated clinical phenotypes such as neurodevelopmental delay or dysmorphic body features.[2] Key features that can differentiate monogenic obesity from polygenic obesity include hyperphagia, extreme symptoms of obesity and the early onset of obesity.[1]
Monogenic obesity is a rare, early-onset form of obesity that follows a Mendelian pattern of inheritance. Most cases arise from mutations in a single gene of the leptin-melanocortin pathway, a key regulatory system controlling body weight, hunger, and energy expenditure.[3] Research on monogenic obesity has focused on characterizing its clinical features, establishing diagnostic criteria, and developing therapeutic approaches, with case studies across diverse populations to improve understanding and management of the condition.
History of Monogenic Obesity
[edit]
The genetic basis of monogenic obesity was first established in 1994 by Jeffery Friedman and colleagues. The discovery of the leptin encoding obese gene (Ob), which established genetic factors as a crucial influence for obesity, allowed for a series of corresponding discoveries that expanded on the influence of single-mutated genes in obesity.[4]
In 1997, two separate reported cases of monogenic obesity were published. Carl Montague and colleagues, and Robert Jackson and colleagues observed severe obesity phenotypes associated with single gene mutations involving the leptin protein/leptin pathway.[4] Their findings established that single genes existed that could directly influence severe human obesity when mutated.
It should be noted that the major contribution of our current understanding of monogenic human obesity is aided by mouse models. Most murine genes identified through monogenic mutation studies have their corresponding human homologs. [1]
Overview of Leptin-Melanocortin pathway
[edit]

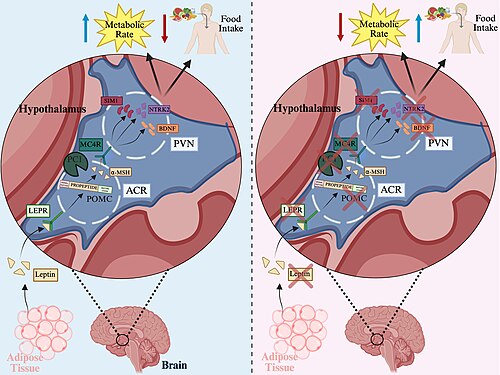
In this pathway, leptin, a hormone secreted by adipose tissue, binds to the leptin receptor in the arcuate nucleus of the hypothalamus. This interaction stimulates the production of proopiomelanocortin (POMC), which is cleaved by prohormone convertase 1 (PC1) to produce α-melanocyte-stimulating hormone (α-MSH). α-MSH then binds to and activates the melanocortin-4 receptor (MC4R) on neurons in the paraventricular nucleus of the hypothalamus, initiating a signaling cascade that ultimately reduces food intake and increases energy expenditure[5]
Leptin is an adipocyte-specific secreted hormone from the obese gene (ob), otherwise known as the leptin gene (LEP). It is a principal regulator of fat mass and is integral in the regulation of fuel stores and energy balance, while signalling the body’s nutritional status to peripheral tissues for modulated functions.[6] Through mice, studies have alluded that leptin commonly regulates many peripheral tissues in humans, such as a mitogenic influence on CD4+ T cells, the circulation of thyrotropin-Releasing Hormone (TRH) that regulates metabolism, the regulation of the onset of puberty, or the neuroendocrine restriction responses to food.[6][7]
Leptin deficiency is a result of a homozygous recessive mutation. In a case study of a Turkish family with obese family members, the homozygous mutation of the leptin gene (LEP) resulted in a C to T substitution, resulting in an Arg to Trp replacement in codon 105.[8] Another case study of two Pakistani children suffering from obesity at a young age was a result of a frameshift mutation on the obese gene (ob), resulting in the deletion of a guanine nucleotide of codon 133.[9] In the case of both families, other family members who were heterozygous for the mutation or homozygous for the WT allele did not display any phenotypes associated with obesity. [8][9]
Leptin deficiency resulted in a range of symptoms including increased body mass index, infertility, delayed onset of puberty, cold intolerance, lower energy expenditures and intense hyperphagia.[1][9][8][6] Increased body mass index was observed in mice and the Pakistani and Turkish case studies, where mice grew to three times the size of the wild type and subjects were born at normal weight, then rapidly increased in weight at an early age.[9][8][6] Initially, infertility due to leptin deficiency was not established, as case studies such as the Pakistani family consisted only of young children. However, infertility was observed in the Turkish family as the adult subjects had hypothalamic-pituitary hormone insufficiency due to leptin deficiency and had never gone into puberty.[8] Cold intolerance was also observed in both the mice and the Turkish subjects when exposed to cold conditions.[8][6] Lastly, a consistent symptom of leptin deficiency across mice and subjects in many case studies is intense hyperphagia and lower energy expenditures.[1][6][8][9]
Subcutaneous recombinant human leptin therapy is a prevalent treatment option for leptin-deficient patients. Positive results from the therapy are observed, including an intense loss of fat mass through decreasing food consumption, an increase in basal metabolic activity and lower serum insulin/cholesterol levels.[1][10] An increase in the proportion of CD4+ T cells is also observed.[10] Another study suggests that leptin therapy has the greatest impact on decreased food intake, while other factors, such as energy expenditure, do increase but do not play as much of a role.[1]
The leptin receptor (LEPR) is a transmembrane protein with an extracellular domain that binds leptin. Its function is mediated by leptin, regulating energy stores, hypothalamic function, and neuroendocrine signaling.[11][12][13](. Mutational analysis demonstrates that a functional leptin receptor is essential for proper regulation of body weight, sexual maturation, and endocrine hormone secretion.[12]
Mutations that produce a secreted, truncated form of the receptor—lacking transmembrane and intracellular domains—are defective in signal transduction but may still bind leptin. In these cases, the mutant receptor in serum can trap leptin, leading to elevated total leptin levels.[13]
In terms of the clinical phenotype of leptin-receptor deficiency, individuals with early-onset obesity with a homozygous loss-of-function mutation in the LEPR all exhibited obesity, hyperphagia, and increased food intake from an early age[11]. However, there was no major deficit in energy expenditure. Vertical growth and IGF-1 levels during childhood were norma,l but adult height was reduced due to a lack of pubertal growth spurt[11][12]. Adults exhibited hypogonadism and reduced secondary sex characteristics, along with low sex-steroid levels, FSH, and LH hormone levels)[11][12]. However, uterine development in adult females was observed, and hormonal levels during the follicular phase were still within the normal range, suggesting that activation of the hypothalamic-pituitary gland was still possible, albeit delayed. Heterozygotes undergo puberty normally and demonstrate normal reproductive functions[12]. Subjects also had reduced CD4+ T cells, but in compensation had higher amounts of B lymphocytes. Surprisingly, the overall clinical phenotype of subjects with leptin receptor deficiency is not as severe as those with leptin deficiency, despite similar phenotypes observed in mice harboring mutations in leptin or its receptor[11]
Melanocortin-4 receptor (MC4R)
[edit]
The melanocortin-4 receptor (MC4R) is a 322-amino acid protein essential for the central regulation of long-term energy. Central regulation consists of regulating food intake and integrating a satiety signal provided to the MC4R from the agonist α-MSH and an orexigenic signal provided by its antagonist agouti-related protein (AGRP).[14][15] MC4R is coupled to a G-protein in the paraventricular nucleus of the hypothalamus, and when activated, it transduces a signal to regulate appetite and decrease or increase food intake.[14][16]
Mutations to the MC4R are the most frequent cause of severe monogenic obesity.[16][17] MC4R mutation can be codominant, and rarer homozygous mutants result in more severe obesity symptoms than heterozygote carriers.[1][14][16][15] There are variations of mutations that can occur and are continuously studied to differentiate between the severity and phenotypes of each mutation. A case study of 289 obese Czech children identified a novel missense mutation, Cys84Arg, as well as 5 previously reported variants: Arg7Cys, Ser19fsdelA, Phe51Leu, Ser127Leu, and Gly181Asp.[16] Further, another case study with 300 Chinese children (200 obese and 100 unaffected) also found novel heterozygous non-synonymous (Val166Ile; Arg310Lys) and nonsense (Cys277Stop) mutations in obese Chinese children.[18] Depending on the type of mutation, this can result in the partial or complete loss of function of MC4R as well as influence the intracellular retention of the MC4R. Intracellular retention appears to provide the most severe obesity phenotypes and earlier age onset, even in comparison to complete loss of function mutations.[16]
MC4R deficiency has common symptoms that involve early-onset obesity, increased bone mineral density, hyperinsulinemia, hyperphagia and increased linear growth in children.[1][17][19] However, it has been noted through many case studies that, depending on the variation of mutation, common symptoms are often not observed.[15][17]
Pro-opiomelanocortin (POMC)
[edit]
POMC is a precursor protein that upon proteolytic cleavage yields melanocortin peptides including adrenocorticotrophin (ACTH), melanocyte-stimulating hormones (MSH), and opioid-receptor ligand β-endorphin. These peptides serve diverse roles such as controlling adrenal growth and skin pigmentation through binding to their corresponding melanocortin receptors (MCRs)[20][21]. Hypothalamic MSH binds to and activates MC3R and MC4R to regulate body weight, pituitary-derived ACTH interacts with the adrenal MC2 receptor to regulate cortisol secretion, and paracrine MSH peptides bind to MC1 receptor to regulate coat pigmentation[22]
The first two reported cases of POMC deficiency involved a patient who was a compound heterozygote for two exon mutations (G7013T in the paternal allele and C7133∆ in the maternal allele), and another patient who was homozygous for a mutation in exon 2 (C3804A) that abolished translation of POMC[20]. Three additional cases in European children included a male patient from Slovenia with decreased adrenal gland size and very low serum cortical and ACTH levels, a Dutch male diagnosed with central hypocortisolism, and a Swiss female diagnosed with hypoglycemia due to ACTH deficiency[22] Another reported case of human POMC deficiency involved a homozygous deletion mutation (C6906del) resulting in the loss of all POMC-derived peptides[21].
Phenotypic Effects:
[edit]
Complete loss-of-function mutations of POMC results in the clinical phenotype of complete POMC deficiency with non-detectable levels of plasma cortisol and ACTH, early-onset and severe obesity, and hypercorticolism[20][21][22]. All patients developed extreme obesity within the first year of life with reports of hyperphagic behavior. Five of the six European patients exhibited red hair, whereas the Turkish patient had brown hair. This difference suggests that darker-haired ethnic groups may carry other genetic variants that maintain eumelanin synthesis in the absence of POMC-derived ligands, while lighter-haired northern European groups may depend more heavily on such ligands[20][21][22].
Significant higher prevalence of obesity observed in heterozygote carriers compared to wild type suggests that loss of one POMC allele is sufficient to increase risk of obesity[22].
As life-threatening symptoms from secondary hydrocortisonemia can appear as early as during the neonatal period of POMC-deficient patients, there is a time window for timely intervention with hydrocortisone replacement therapy. Otherwise, fatality may result as seen in a first-born son of a family with POMC deficiency who died because of hepatic failure before hypocortisolism could be diagnosed[21].
Prohormone convertase 1/3 (PC1/3)
[edit]
Prohormone convertase 1/3 (PC1/3) is involved in the regulation of energy balance through leptin. Several proneuropeptides/prohormones, such as proTRH, pro-opiomelanocortin, proinsulin, and progonadotropin, require cleavage at the C-terminal basic residues.[23][1] Carboxypeptidase E (Cp3) is responsible for the prohormone/propeptide cleavage at the C-terminal. The post-translational cleavage regulation required for energy balance relies on PC1/3. Studies hypothesize that PC1/3 works upstream of Cpe to regulate its cleavage activity and activate proneurohormones into prominent mature proteins that work in the anterior pituitary, hypothalamus and/or skin.[5][24]
PC1/3 deficiency is a result of autosomal recessive obesity. An example case study observed a woman with two different mutant alleles with the mutations Gly→Arg483 leading to ER retention failure and A→C+4 of the intron-5 donor splice site, causing a frameshift and creation of a premature stop codon within the catalytic domain.[24] Mutations in PC1/3 prevent the critical post-translational regulation, thus resulting in impaired prohormone processing that has many downstream impacts affecting energy regulation. It is important to note that very few cases of PC1/3 deficiency are reported, and often, obesity is not the most prominent phenotype, suggesting that PC1/3 deficiency is rare in diagnosing monogenic obesity.
Phenotypic Effects:
[edit]
PC1/3 deficiency has several syndromes, including childhood-onset obesity, hypoglycemia, hypogonadotropic hypogonadism, severe postnatal growth, and elevated proinsulin, all due to impaired post-transplantation prohormone processing of key regulatory genes.[25]
Single-minded homolog 1 (S1M1)
[edit]
SIM1 is a transcription factor that plays an essential role in the formation of the paraventricular nucleus (PVN) of the hypothalamus in mice, which integrates signals related to appetite and energy expenditure[26]. It is a mammalian homolog of the Drosophila transcription factor Single-minded, belonging to the bHLH-PAS family. In Drosophila, homozygous loss of Single-minded results in the failure to form midline central nervous system structures. SIM1 is strongly expressed in the PVN, and studies of its mouse homologs SIM1 and SIM2 indicate that these genes are important for neuroendocrine function by regulating the terminal differentiation of several neuronal cell types[27].
A study of an 18-month-old girl reported a de novo balanced translocation between the short arm of chromosome 1 and the long arm of chromosome 7, disrupting the SIM1 gene on 6q while leaving the transcription unit on 1p unaffected. She carried a C→T substitution in one SIM1 allele, indicating haploinsufficiency and heterozygosity for a loss-of-function mutation. This suggested that obesity caused by SIM1 mutations follows an autosomal dominant pattern of inheritance[26].
In mice, homozygous Sim1 knockout results in prenatal lethality due to failure of PVN development, while heterozygous mice survive but develop early-onset obesity, increased linear growth, hyperinsulinemia, hyperleptinemia, and hyperphagia without a corresponding decrease in energy expenditure[27].
In humans, a de novo balanced translocation between chromosomes 1 and 7 has been associated with severe obesity and increased linear growth, with a rate of weight gain comparable to that seen in patients with leptin or leptin receptor mutations, alongside elevated serum leptin concentrations[26]. These findings parallel those from Sim1 heterozygous mice, where hyperphagia in the absence of decreased energy expenditure suggests that obesity is primarily driven by increased food intake.
A 21-month-old male with a novel SIM1 mutation presented with early-onset obesity and hypopituitarism following endocrine evaluation[28]. The mutation was maternally inherited, but the mother exhibited a milder, later-onset obesity phenotype, suggesting variable penetrance due to additional genetic or environmental factors influencing SIM1-related pathology.
In cases of hypopituitarism, treatment with levothyroxine, hydrocortisone, and desmopressin is required to manage pituitary hormone deficiencies and maintain hormone levels within the normal range[28].
Brain-Derived Neurotrophic Factor (BDNF)
[edit]
Brain-derived neurotrophic factor (BDNF) belongs to the neurotrophin family. The neurotrophin is synthesized as a prohormone that is then cleaved by prohormone convertase.[29] Studies of adult rat brains show that BDNF mRNA is commonly expressed in the hippocampus, septum, cortex and the hypothalamic nuclei associated with satiety and locomotor activities.[30] Studies infer that the location of BDNF may enter neurons to directly influence transcription as well as control eating behaviour through signals of satiety, inhibiting food intake.[29] Therefore, the role of BDNF is to maintain energy homeostasis through regulating food intake as well as cognitive functions, memory and behaviour, likely working downstream of MC4R.[1][31]
Studies of mice show that when BDNF is mutated, the disrupted neurotrophin results in increased food intake and obesity in mice.[30][32] Further, when various heterozygous mice had targeted disruption of proteins involved in the neurotrophin pathway, such as BDNF, NT-4/5 or TrkA, only BDNF mutants had significant increases in weight compared to WT.[30] A case study with an eight-year-old girl displayed a chromosomal inversion in the BDNF gene, displaying a common polymorphism. When compared to other individuals of the same age and BMI, the girl displayed reduced serum BDNF levels and gained weight much faster than others, becoming obese at the age of eight.[1][31]
Phenotypic Effects:
[edit]
Common symptoms associated with BDNF mutations involved intense hyperphagia, severe obesity, impaired cognitive functions such as memory and nociception and hyperactivity.[1][30][32]
Various therapeutic approaches are under research involving stimulating the transcription of BDNF or restoring BDNF levels, but there has been no clear therapeutic solution yet.[33] A study has hypothesized that infusion with BDNF may reverse the inability to regulate food intake and prevent obesity, yet the results are only transient.[30]
Neurotrophic Receptor Tyrosine Kinase 2 (NTRK2)
[edit]
NTRK2 is part of the TRK family of receptor tyrosine kinases that are activated by neurotrophins. Neurotrophin signaling through these receptors regulate various functions such as cell survival and proliferation, with the NTRK2-BDNF interaction specifically serving important roles in regulating mammalian eating behavior and energy balance[34][35]. TRKs contain cytoplasmic domains that have phosphorylation sites capable of recruiting intermediates for further signal transduction. Infusing MC4R deficient mice with BDNF suppressed the increased food intake and weight gain, suggesting that NTRK2 acts downstream of MC4R[35].
A child with severe early onset obesity and hyperphagia was found to be a heterozygote for a de novo loss of function mutation in NTRK2 where an A to G transition occurred in codon 722[35].
Despite normal birth weight, the child harboring the Y722DC mutation gained weight rapidly from an age of 6 months. He was also reported to be hyperphagic and also exhibited increased linear growth throughout childhood[35]. The Y722C mutation impaired ligand-induced phosphorylation and impairment of MAPK phosphorylation, a signaling pathway downstream of the receptor[35].
A subsequent functional characterization of the Y622C mutation showed that in addition to loss of signaling, this mutation impairs the ability of NTRK2 to promote neurite outgrowth in response to the addition of BDNF. This suggests that the severe hyperphagic and obesity seen in this individual can be mechanistically explained by impaired hypothalamic neurogenesis[36]
- ^ a b c d e f g h i j k l m Chung, Wendy K. (2012-01). “An overview of mongenic and syndromic obesities in humans”. Pediatric Blood & Cancer. 58 (1): 122–128. doi:10.1002/pbc.23372. ISSN 1545-5017. PMC 3215910. PMID 21994130.
- ^ Fitch, Angela K.; Malhotra, Sonali; Conroy, Rushika (2024-09-01). “Differentiating monogenic and syndromic obesities from polygenic obesity: Assessment, diagnosis, and management”. Obesity Pillars. 11: 100110. doi:10.1016/j.obpill.2024.100110. ISSN 2667-3681.
{{cite journal}}: CS1 maint: article number as page number (link) - ^ Sohn, Young Bae (2022-12-29). “The genetics of obesity: A narrative review”. Precision and Future Medicine. 6 (4): 226–232. doi:10.23838/pfm.2022.00156. ISSN 2508-7940.
- ^ a b Kincaid, John W. R. (2023-07). “The discovery of human monogenic obesity”. Nature Reviews Endocrinology. 19 (7): 381–381. doi:10.1038/s41574-023-00844-1. ISSN 1759-5037.
- ^ a b Ranadive, Sayali A.; Vaisse, Christian (2008-09). “Lessons from extreme human obesity: monogenic disorders”. Endocrinology and Metabolism Clinics of North America. 37 (3): 733–751, x. doi:10.1016/j.ecl.2008.07.003. ISSN 0889-8529. PMC 5877402. PMID 18775361.
- ^ a b c d e f Friedman, J. M.; Halaas, J. L. (1998-10-22). “Leptin and the regulation of body weight in mammals”. Nature. 395 (6704): 763–770. doi:10.1038/27376. ISSN 0028-0836. PMID 9796811.
- ^ Gibson, William T.; Farooqi, I. Sadaf; Moreau, Mary; DePaoli, Alex M.; Lawrence, Elizabeth; O’Rahilly, Stephen; Trussell, Rebecca A. (2004-10). “Congenital leptin deficiency due to homozygosity for the Delta133G mutation: report of another case and evaluation of response to four years of leptin therapy”. The Journal of Clinical Endocrinology and Metabolism. 89 (10): 4821–4826. doi:10.1210/jc.2004-0376. ISSN 0021-972X. PMID 15472169.
- ^ a b c d e f g Strobel, A.; Issad, T.; Camoin, L.; Ozata, M.; Strosberg, A. D. (1998-03). “A leptin missense mutation associated with hypogonadism and morbid obesity”. Nature Genetics. 18 (3): 213–215. doi:10.1038/ng0398-213. ISSN 1061-4036. PMID 9500540.
- ^ a b c d e Montague, C. T.; Farooqi, I. S.; Whitehead, J. P.; Soos, M. A.; Rau, H.; Wareham, N. J.; Sewter, C. P.; Digby, J. E.; Mohammed, S. N.; Hurst, J. A.; Cheetham, C. H.; Earley, A. R.; Barnett, A. H.; Prins, J. B.; O’Rahilly, S. (1997-06-26). “Congenital leptin deficiency is associated with severe early-onset obesity in humans”. Nature. 387 (6636): 903–908. doi:10.1038/43185. ISSN 0028-0836. PMID 9202122.
- ^ a b Farooqi, I. Sadaf; Matarese, Giuseppe; Lord, Graham M.; Keogh, Julia M.; Lawrence, Elizabeth; Agwu, Chizo; Sanna, Veronica; Jebb, Susan A.; Perna, Francesco; Fontana, Silvia; Lechler, Robert I.; DePaoli, Alex M.; O’Rahilly, Stephen (2002-10-15). “Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency”. The Journal of Clinical Investigation. 110 (8): 1093–1103. doi:10.1172/JCI15693. ISSN 0021-9738. PMID 12393845.
- ^ a b c d e Farooqi, I. Sadaf; Wangensteen, Teresia; Collins, Stephan; Kimber, Wendy; Matarese, Giuseppe; Keogh, Julia M.; Lank, Emma; Bottomley, Bill; Lopez-Fernandez, Judith; Ferraz-Amaro, Ivan; Dattani, Mehul T.; Ercan, Oya; Myhre, Anne Grethe; Retterstol, Lars; Stanhope, Richard (2007-01-18). “Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor”. The New England Journal of Medicine. 356 (3): 237–247. doi:10.1056/NEJMoa063988. ISSN 1533-4406. PMC 2670197. PMID 17229951.
- ^ a b c d e Clément, K.; Vaisse, C.; Lahlou, N.; Cabrol, S.; Pelloux, V.; Cassuto, D.; Gourmelen, M.; Dina, C.; Chambaz, J.; Lacorte, J. M.; Basdevant, A.; Bougnères, P.; Lebouc, Y.; Froguel, P.; Guy-Grand, B. (1998-03-26). “A mutation in the human leptin receptor gene causes obesity and pituitary dysfunction”. Nature. 392 (6674): 398–401. doi:10.1038/32911. ISSN 0028-0836. PMID 9537324.
- ^ a b Lahlou, N.; Clement, K.; Carel, J. C.; Vaisse, C.; Lotton, C.; Le Bihan, Y.; Basdevant, A.; Lebouc, Y.; Froguel, P.; Roger, M.; Guy-Grand, B. (2000-08). “Soluble leptin receptor in serum of subjects with complete resistance to leptin: relation to fat mass”. Diabetes. 49 (8): 1347–1352. doi:10.2337/diabetes.49.8.1347. ISSN 0012-1797. PMID 10923636.
- ^ a b c Lubrano-Berthelier, Cécile; Le Stunff, Catherine; Bougnères, Pierre; Vaisse, Christian (2004-05). “A homozygous null mutation delineates the role of the melanocortin-4 receptor in humans”. The Journal of Clinical Endocrinology and Metabolism. 89 (5): 2028–2032. doi:10.1210/jc.2003-031993. ISSN 0021-972X. PMID 15126516.
- ^ a b c Lubrano-Berthelier, Cecile; Dubern, Beatrice; Lacorte, Jean-Marc; Picard, Franck; Shapiro, Astrid; Zhang, Sumei; Bertrais, Sandrine; Hercberg, Serge; Basdevant, Arnaud; Clement, Karine; Vaisse, Christian (2006-05). “Melanocortin 4 receptor mutations in a large cohort of severely obese adults: prevalence, functional classification, genotype-phenotype relationship, and lack of association with binge eating”. The Journal of Clinical Endocrinology and Metabolism. 91 (5): 1811–1818. doi:10.1210/jc.2005-1411. ISSN 0021-972X. PMID 16507637.
- ^ a b c d e Hainerová, Irena; Larsen, Lesli H.; Holst, Birgitte; Finková, Marie; Hainer, Vojtech; Lebl, Jan; Hansen, Torben; Pedersen, Oluf (2007-09). “Melanocortin 4 receptor mutations in obese Czech children: studies of prevalence, phenotype development, weight reduction response, and functional analysis”. The Journal of Clinical Endocrinology and Metabolism. 92 (9): 3689–3696. doi:10.1210/jc.2007-0352. ISSN 0021-972X. PMID 17579204.
- ^ a b c Farooqi, I. Sadaf; Keogh, Julia M.; Yeo, Giles S. H.; Lank, Emma J.; Cheetham, Tim; O’Rahilly, Stephen (2003-03-20). “Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene”. The New England Journal of Medicine. 348 (12): 1085–1095. doi:10.1056/NEJMoa022050. ISSN 1533-4406. PMID 12646665.
- ^ Wang, C. L.; Liang, L.; Wang, H. J.; Fu, J. F.; Hebebrand, J.; Hinney, A. (2006-11). “Several mutations in the melanocortin 4 receptor gene are associated with obesity in Chinese children and adolescents”. Journal of Endocrinological Investigation. 29 (10): 894–898. doi:10.1007/BF03349193. ISSN 1720-8386. PMID 17185898.
- ^ Dubern, Béatrice; Bisbis, Selma; Talbaoui, Habiba; Le Beyec, Johanne; Tounian, Patrick; Lacorte, Jean-Marc; Clément, Karine (2007-06). “Homozygous null mutation of the melanocortin-4 receptor and severe early-onset obesity”. The Journal of Pediatrics. 150 (6): 613–617, 617.e1. doi:10.1016/j.jpeds.2007.01.041. ISSN 1097-6833. PMID 17517245.
- ^ a b c d Krude, H.; Biebermann, H.; Luck, W.; Horn, R.; Brabant, G.; Grüters, A. (1998-06). “Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans”. Nature Genetics. 19 (2): 155–157. doi:10.1038/509. ISSN 1061-4036. PMID 9620771.
- ^ a b c d e Farooqi, I. Sadaf; Drop, Stenvert; Clements, Agnes; Keogh, Julia M.; Biernacka, Joanna; Lowenbein, Sarah; Challis, Benjamin G.; O’Rahilly, Stephen (2006-09). “Heterozygosity for a POMC-null mutation and increased obesity risk in humans”. Diabetes. 55 (9): 2549–2553. doi:10.2337/db06-0214. ISSN 0012-1797. PMID 16936203.
- ^ a b c d e Krude, Heiko; Biebermann, Heike; Schnabel, Dirk; Tansek, Mojca Zerjav; Theunissen, Pierre; Mullis, Primus E.; Grüters, Annette (2003-10). “Obesity due to proopiomelanocortin deficiency: three new cases and treatment trials with thyroid hormone and ACTH4-10”. The Journal of Clinical Endocrinology and Metabolism. 88 (10): 4633–4640. doi:10.1210/jc.2003-030502. ISSN 0021-972X. PMID 14557433.
- ^ Nillni, Eduardo A. (2007-09). “Regulation of prohormone convertases in hypothalamic neurons: implications for prothyrotropin-releasing hormone and proopiomelanocortin”. Endocrinology. 148 (9): 4191–4200. doi:10.1210/en.2007-0173. ISSN 0013-7227. PMID 17584972.
- ^ a b Jackson, R. S.; Creemers, J. W.; Ohagi, S.; Raffin-Sanson, M. L.; Sanders, L.; Montague, C. T.; Hutton, J. C.; O’Rahilly, S. (1997-07). “Obesity and impaired prohormone processing associated with mutations in the human prohormone convertase 1 gene”. Nature Genetics. 16 (3): 303–306. doi:10.1038/ng0797-303. ISSN 1061-4036. PMID 9207799.
- ^
- ^ a b c Holder, J. L.; Butte, N. F.; Zinn, A. R. (2000-01-01). “Profound obesity associated with a balanced translocation that disrupts the SIM1 gene”. Human Molecular Genetics. 9 (1): 101–108. doi:10.1093/hmg/9.1.101. ISSN 0964-6906. PMID 10587584.
- ^ a b Michaud, J. L.; Boucher, F.; Melnyk, A.; Gauthier, F.; Goshu, E.; Lévy, E.; Mitchell, G. A.; Himms-Hagen, J.; Fan, C. M. (2001-07-01). “Sim1 haploinsufficiency causes hyperphagia, obesity and reduction of the paraventricular nucleus of the hypothalamus”. Human Molecular Genetics. 10 (14): 1465–1473. doi:10.1093/hmg/10.14.1465. ISSN 0964-6906. PMID 11448938.
- ^ a b Gonsalves, Rob; Aleck, Kirk; Newbern, Dorothee; Shaibi, Gabriel; Kapadia, Chirag; Oatman, Oliver (2020-10-06). “Severe early onset obesity and hypopituitarism in a child with a novel SIM1 gene mutation”. Endocrinology, Diabetes & Metabolism Case Reports. 2020: 20–0042, EDM200042. doi:10.1530/EDM-20-0042. ISSN 2052-0573. PMC 7576654. PMID 33434169.
- ^ a b Tapia-Arancibia, Lucia; Rage, Florence; Givalois, Laurent; Arancibia, Sandor (2004-07). “Physiology of BDNF: focus on hypothalamic function”. Frontiers in Neuroendocrinology. 25 (2): 77–107. doi:10.1016/j.yfrne.2004.04.001. ISSN 0091-3022. PMID 15571756.
- ^ a b c d e Kernie, S. G.; Liebl, D. J.; Parada, L. F. (2000-03-15). “BDNF regulates eating behavior and locomotor activity in mice”. The EMBO journal. 19 (6): 1290–1300. doi:10.1093/emboj/19.6.1290. ISSN 0261-4189. PMC 305670. PMID 10716929.
- ^ a b Gray, Juliette; Yeo, Giles S. H.; Cox, James J.; Morton, Jenny; Adlam, Anna-Lynne R.; Keogh, Julia M.; Yanovski, Jack A.; El Gharbawy, Areeg; Han, Joan C.; Tung, Y. C. Loraine; Hodges, John R.; Raymond, F. Lucy; O’rahilly, Stephen; Farooqi, I. Sadaf (2006-12). “Hyperphagia, severe obesity, impaired cognitive function, and hyperactivity associated with functional loss of one copy of the brain-derived neurotrophic factor (BDNF) gene”. Diabetes. 55 (12): 3366–3371. doi:10.2337/db06-0550. ISSN 0012-1797. PMC 2413291. PMID 17130481.
- ^ a b Pelleymounter, M. A.; Cullen, M. J.; Wellman, C. L. (1995-02). “Characteristics of BDNF-induced weight loss”. Experimental Neurology. 131 (2): 229–238. doi:10.1016/0014-4886(95)90045-4. ISSN 0014-4886. PMID 7534721.
- ^ Rosas-Vargas, Haydeé; Martínez-Ezquerro, José Darío; Bienvenu, Thierry (2011-08-01). “Brain-Derived Neurotrophic Factor, Food Intake Regulation, and Obesity”. Archives of Medical Research. 42 (6): 482–494. doi:10.1016/j.arcmed.2011.09.005. ISSN 0188-4409.
- ^ Huang, Eric J.; Reichardt, Louis F. (2003-06). “Trk Receptors: Roles in Neuronal Signal Transduction”. Annual Review of Biochemistry. 72 (1): 609–642. doi:10.1146/annurev.biochem.72.121801.161629. ISSN 0066-4154.
- ^ a b c d e Yeo, Giles S H; Connie Hung, Chiao-Chien; Rochford, Justin; Keogh, Julia; Gray, Juliette; Sivaramakrishnan, Shoba; O’Rahilly, Stephen; Farooqi, I Sadaf (2004-11). “A de novo mutation affecting human TrkB associated with severe obesity and developmental delay”. Nature Neuroscience. 7 (11): 1187–1189. doi:10.1038/nn1336. ISSN 1097-6256.
- ^ Gray, J; Yeo, G; Hung, C; Keogh, J; Clayton, P; Banerjee, K; McAulay, A; O’Rahilly, S; Farooqi, I S (2007-02). “Functional characterization of human NTRK2 mutations identified in patients with severe early-onset obesity”. International Journal of Obesity. 31 (2): 359–364. doi:10.1038/sj.ijo.0803390. ISSN 0307-0565.



